
Evolution in Court
|
The last two decades have witnessed the widespread use of genetic testing in courtrooms, to identify criminals from biological remains, determine paternity or identify corpses. Less is known, however, about expert reports based on the use of molecular phylogenies. Such reports are founded on studies into the common ancestry of organisms, usually bacteria or viruses, to trace them back to the source of infection. Such cases range from a Florida dentist who infected several of his patients with HIV, to a Spanish anaesthetist, Juan Maeso, who transmitted hepatitis C virus (HCV) to almost 300 patients. Evolutionary theory plays an essential role in resolving cases like these, and this article explains how and why evolutionary theory is pertinent. Keywords: common ancestor, virus, molecular clock, likelihood ratio, evolutionary hypotheses. |
||
|
Scientific theories are not tried in the courtroom, or at least they have not been for several centuries. When Galileo Galilei, Giordano Bruno or Miguel Servet, among others, were put on trial, they were tried in ecclesiastical courts, not civil courts, and mostly they were convicted based on religious clashes and not necessarily due to heresy arising from new scientific theories. There is one notable exception: the theory of biological evolution, formulated by Charles Darwin and Alfred R. Wallace in the mid nineteenth century. Evolutionary theory is regularly questioned in U.S. courts at all levels, which have shed doubt on its validity as a scientific theory worthy of inclusion on secondary education curricula. But here we are concerned with its presence in the courtroom in quite another context. Evolution has been appearing in court since the 1990s. In a growing number of cases, evolutionary biologists are called to testify as expert witnesses in civil and, occasionally, criminal lawsuits. There is a common thread joining them all: the transmission of an infectious agent, usually a virus such as human immunodeficiency virus (HIV) or hepatitis C virus (HCV) from a common source to one or more infected recipients. Given the severity of these infections, which can lead to the patient’s death, people generally seek compensation in court for physical, psychological and economic damages. Frequently the source is related to a medical procedure, a transfusion with contaminated blood or another substance, or the use of an unsterilized product or equipment, but we also find cases of malpractice, even criminal acts. Evolution speeded up One of the main tenets of the theory of evolution is the common ancestry of all living beings. Although some of the finer details are still controversial regarding the early stages of life –once cells had formed– the evidence in favour of the common origin of all organisms is strong enough for this idea to be accepted by current biology as one of its pillars. Accordingly, and since one of the basic principles establishes physical continuity between the hereditary component of ancestors and their descendants, between parents (mainly mothers, when there is sexual differentiation) and offspring, it would theoretically be possible to draw a giant tree linking all living things that exist, or have existed and left descendants to this day, by studying their hereditary material. There are several projects with similar, albeit less ambitious objectives, endeavouring to unravel what is known as the «Tree of Life». The methods used are primarily based on analysing the variation and evolution of hereditary material, mainly DNA (deoxyribonucleic acid), which is the common inheritance molecule of all cellular organisms. Some viruses, however, including important pathogens such as HIV or influenza, have another molecule, RNA (ribonucleic acid), to transmit the genetic message. Molecular phylogenetics can establish kinship between all types of organisms exactly and reliably by using increasingly complex models. To do so, the processes of evolutionary change are represented at the molecular level using sophisticated computational methods, comparing the nucleotide sequences bearing the hereditary message. Unlike other applications of biology in forensic analysis, which seek to establish a link between the biological remains found at a crime scene and possible suspects, or to determine kinship between individuals, the forensic application of phylogenetic sequence analysis is based on studying the cumulative differences between sequences since the time they diverged from the common ancestor. Throughout evolution, we consider that different lineages arising from the differentiation a common ancestor will accumulate differences at all levels. These differences range from the molecular to the behavioural and accumulate independently so, over generations, what was initially an almost perfect identity leads to an increasing number of differences. Broadly speaking, a greater number of accumulated differences will indicate that a greater amount of time has elapsed since the compared organisms diverged from their common ancestor. The number of differences observed depends on two basic factors, time and the rate of evolutionary change, which can only be separated in principle from independent estimates of one or the other. Depending on the type of organism, on the type of traits compared, and on the evolutionary rate associated with them, there are cases where there is little difference after millions of years of independent evolution, while in others, just a few days or weeks is long enough to appreciate differences. Organisms that exhibit such rapid rates of evolutionary change often have RNA-based genomes and, as already indicated, tend to be pathogens such as HIV, HCV or the influenza A virus. Their evolutionary rates are up to one million times faster than those of the organisms they infect. This high speed is the result of their peculiar machinery for copying hereditary material, which cannot correct copy errors, and of the huge population sizes (billions of new virus particles are produced every day in an infected patient), as well as a very short generation time, lasting just a few minutes. This high evolutionary rate has important clinical consequences, such as making it difficult to develop effective vaccines for HIV or HCV, the rapid development of mutants that are resistant to antiviral treatment, or the ease with which they adapt to new hosts. On the other side of the coin, however, we can see how their populations evolve in real time because enough changes accumulate over weeks or months to enable the application of molecular phylogenetic methods to reconstruct their short-term history. |
«In a growing number of cases, evolutionary biologists are called to testify as expert witnesses in civil or criminal lawsuits. There is a common thread joining them all: the transmission of an infectious agent» «Molecular phylogenetics aims to reconstruct the evolutionary and genealogical relationships between organisms from homologous proteins or gene sequences» |
|
 2007 Tree of Life Web Project. Image of Rose © 1999 Nick Kurzenko. Image of annelid worm © 2001 Greg W. Rouse 2007 Tree of Life Web Project. Image of Rose © 1999 Nick Kurzenko. Image of annelid worm © 2001 Greg W. RouseOne of the main tenets of the theory of evolution is the common ancestry of all living beings. Accordingly, and since one of the basic principles establishes physical continuity between the hereditary component of ancestors and their descendants, it would theoretically be possible to draw a giant tree linking all living things that exist, or have existed and left descendants to this day, by studying their hereditary material. There are several projects with similar, albeit less ambitious objectives, endeavouring to unravel what is known as the «Tree of Life». |
«HIV infection was diagnosed in a woman who did not engage in any known risk practices, and this led to an investigation that pointed to her HIV-positive dentist as the possible source of infection» |
|
|
molecular phylogeny Molecular phylogenetics sets out to reconstruct the evolutionary and genealogical relationships between organisms from homologous proteins or gene sequences. This scientific discipline dates back to the mid nineteen sixties, but it has developed very rapidly early in this century, with the accumulation of data on gene and genome sequences from diverse organisms. Also, the development of new algorithms and the increasing computational power of computers have led to the application of more sophisticated methods, such as maximum likelihood or Bayesian inference, which can deal with ever larger data sets. The result is that nowadays we have procedures to construct and contrast increasingly complex evolutionary hypotheses. When applied to cases of infections with rapidly evolving pathogens, molecular phylogenies are an essential tool to unravel possible transference and direction (who infects who), the sources of outbreaks, estimate when infection occurred, etc., applied to events separated by a few weeks or months. When molecular phylogenetics is applied to court cases, a basic question to resolve is whether samples obtained from different patients share a common ancestor that is closer to the possible source of infection than to other infected individuals in the general population, considered as external controls. The first case in which this methodology was applied is an excellent illustration of the problem and how it was solved. Infections and court cases In the late nineteen eighties, the AIDS epidemic spread unchecked through many different countries and communities. Then there were no drugs available to slow down the progression of AIDS and its diagnosis represented little more than the announcement of imminent death. It was known that the disease was caused by a retrovirus and that it was passed from person to person through bodily fluids, especially blood, but could also be transmitted by intermediate vectors, such as hypodermic needles. Then HIV infection was diagnosed in a woman who did not engage in any known risk practices, and this led to an investigation that pointed to her HIV-positive dentist as the possible source of infection. Until that time it was not thought possible for HIV to be transmitted through dental instruments, thus no safety measures were taken. The woman in question decided to take a civil action against her dentist, and sue him for damages. The case became more complicated when it was discovered that six other patients of the same dentist were also infected with HIV. The question now was no longer whether, or not, the first patient had been infected by the dentist, but how many and which patients had been infected by him, as some of them did engage in risk practices. To answer this, an expert team was called in, which sequenced a fragment of the viral genome in samples obtained from both the dentist and his patients, and 35 other people unrelated with them but also infected with HIV. The judge in charge of the case was not competent enough to take a decision on the validity of the evidence presented. Basically, a phylogenetic study linked samples from five patients, including the first petitioner, with the dentist but ruled out the other two, whose source of origin was undetermined. Therefore the following resolution was adopted: the judge would accept the validity of the test if the scientific community ratified it. To do so, the results should be communicated in the usual way, as a scientific article to a peer-reviewed journal, and thus be submitted to the appropriate scrutiny and evaluation before publication. The article was published in the journal Science on May 22, 1992 (Ou et al., 1992) and the dentist was condemned to compensate five of the victims for damages. Unlike other genetic evidence commonly employed in court cases, here it did not seek to establish identity between the alleged victims’ genetic data and that of the source of infection, but rather to distinguish between their common co-ancestry and the possibility that they came from other sources, which were not identified in the general population. All this could be revealed by analysing the differences found between the corresponding gene sequences. Since then, there have been countless cases where we have used the rapid evolution of viruses, like AIDS, to settle claims related to person-to-person infection or infection through medical procedures. On occasion, proceedings have been held in a criminal court, rather than a civil one, as in the case of Richard Schmidt, a physician from Louisiana State (U.S.A.) charged with attempted murder. This American doctor used blood infected with human immunodeficiency virus and hepatitis C virus from one of his patients to transmit these diseases to his former lover, who had decided to break off their relationship (Metzker et al., 2002). Although both viruses were transmitted to the recipient, the prosecution relied on the HIV study to demonstrate the connection. Indeed, numerous advances can be witnessed in the decade separating this case and the case of the Florida dentist, both in terms of the methodology employed and the courts’ acceptance of this evidence. However, it should be mentioned that in both cases doubts were raised about the statistical validation of the results, especially given the importance of the findings. One solution to this problem is the use of statistical methods that are more advanced than the pseudorandom re-sampling (bootstrapping) used until that time. Specifically, the introduction of maximum likelihood estimates of trees means alternative hypotheses can be compared on a solid statistical basis. In fact, two of the authors of reference in forensic genetic applications propose the application of Bayesian inference, based partly on calculating the plausibility of a hypothesis from the available evidence, to report on the likelihood that certain biological elements come from the accused, or not (Evett and Weir, 1998). To do this, the researcher must calculate the probability of observing the data they have under different assumptions as to how these data might have been generated. |
«On occasion, proceedings involve criminal lawsuits. This was the case of Richard Schmidt charged with attempted murder after using HIV and HCV-infected blood to transmit these diseases to his former lover»
|
|
|
|
«It is estimated that about 2.5% of the Spanish population is infected with HCV, although many of those infected are unaware of their status, as the virus may be latent» |
|
|
THE « Maeso casE»: HOW MOLECULAR PHYLOGENETICS SOLVEd A hepatitis C OUTBREAK Our research group has had occasion to employ these procedures in what is probably one of the most complex cases of applying evolutionary theory to expert forensic evidence (González-Candelas et al., 2013). In February 1998, an outbreak of hepatitis C viral infection was discovered in Valencia, which was quickly attributed to the less than negligent work performance of the anaesthetist Dr. Juan Maeso. The so-called «Maeso case» went on for almost 10 years, during which the extensive judicial investigation was followed by a trial in the Provincial Court lasting over a year, with a subsequent appeal to the Supreme Court. As a result the defendant was sentenced to almost 2,000 years in prison for malpractice leading to the infection of at least 275 patients, five of whom had died by the time the trial was held. In this case, the problem was aggravated by the sheer dimension of the outbreak, the largest as yet, which came from the same source over a time period, which had to be determined (it proved to be almost ten years). During this time, the virus had continued to evolve at the source, so it was quite different in the population infected at first from those affected towards the end. The hypothesis on which we based our report was the same as in previous cases: if transmission had occurred from a common source, then the viruses isolated from these patients would share with each other and with the viruses isolated from the source, a nearest common ancestor. This relationship would be closer than with any other viral population not linked to the source. However, in this case we could not just test this hypothesis: it was necessary to establish which of the patients, numbering just under 400, treated by this anaesthetist and diagnosed with HCV infection of the same type, had been infected by him and which ones had been infected from other sources. It is estimated that about 2.5% of the Spanish population is infected with HCV, although many of those infected are unaware of their status as the virus may be latent, without causing apparent disease for years. Notwithstanding it can still be transmitted in this «dormant» state. As in previous studies, we started to sequence viruses from patients who were epidemiologically linked to the outbreak, i.e., those who had been treated by Dr. Maeso in the many hospitals where he practised regularly. Likewise, we sequenced as many viral samples as we could find of the same type but from infected people who were not related to the outbreak. In total, we obtained 4,184 sequence fragments of just over 400 nucleotides, from 366 patients, including 44 controls. The resulting phylogenetic tree (Figure 1) enabled us to establish common ancestry –and therefore inclusion in the outbreak– of sequences from 274 patients, implying the exclusion of 47 others. Furthermore, calculating the likelihood estimates for each case under the two possible hypotheses (that the patient in question belonged to the outbreak or not), it was possible to estimate for each individual the likelihood that the infection was common to the outbreak or not. The analysis was completed with another application of the theory of evolution at the molecular level, based on quantifying the number of changes per unit of time since divergence from a common ancestor. This process occurs at a more or less constant rate, which has led to postulate the existence of a «molecular clock» of evolution (Zuckerkandl and Pauling, 1962). Since its formulation, the molecular clock has been used to date all kinds of evolutionary events (www.timetree.org o www.datelife.org) and recent work means it can even be implemented without an external calibration reference (Drummond et al., 2006). When used with samples derived from the outbreak (Figure 2), this method enabled us to compare the estimated dates of infection from viral sequences with the dates of contagion given by the prosecution, resulting in a high degree of concurrence. During the trial, this assessment was used to determine which insurance companies should pay compensation to each patient, as Dr. Maeso had contracted various insurance policies throughout his professional life. It was also established that the infections had started back in 1988, ten years before the outbreak was detected. Here we have seen how methodological developments in sequencing hereditary material, advances in computational biology and interpretation of the information generated, together with evolutionary theory, which is fundamental for interpreting the data obtained, are all instrumental in shedding light on the origin and timing of the transmission of infectious organisms. Given the civil, and even criminal, responsibilities that can be derived from these infections, we can expect evolution to gain an increasingly prominent role in the courtrooms of the future. |
«These developments are instrumental in shedding light on the origin and timing of the transmission of infectious organisms» |
|
 Figure 1. Phylogenetic tree obtained by maximum likelihood from 4,184 sequence fragments (406 nucleotides) of the hepatitis C virus from an outbreak of the virus detected in Valencia in 1998. The green and red stars indicate the nodes where the nearest common ancestor is located of viruses taken from patients related to the outbreak (green) and the general population (red). The purple and dark blue sequences correspond to control samples and patients excluded from the outbreak, while the remaining sequences correspond to patients from the outbreak, coloured according to the approximate date of infection.
Figure 1. Phylogenetic tree obtained by maximum likelihood from 4,184 sequence fragments (406 nucleotides) of the hepatitis C virus from an outbreak of the virus detected in Valencia in 1998. The green and red stars indicate the nodes where the nearest common ancestor is located of viruses taken from patients related to the outbreak (green) and the general population (red). The purple and dark blue sequences correspond to control samples and patients excluded from the outbreak, while the remaining sequences correspond to patients from the outbreak, coloured according to the approximate date of infection.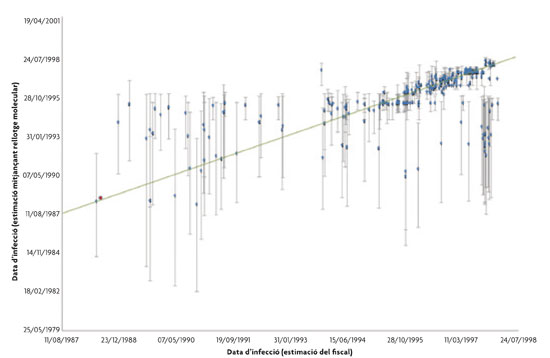 Figure 2. Relationship between the infection-date estimates obtained by hepatitis C virus sequence analysis in patients from the Valencia outbreak, and the prosecutor’s estimates based on patient records. Bars mark intervals of maximum likelihood density at 95% for each estimated date that corresponds to the average obtained through Bayesian analysis for each patient separately.
Figure 2. Relationship between the infection-date estimates obtained by hepatitis C virus sequence analysis in patients from the Valencia outbreak, and the prosecutor’s estimates based on patient records. Bars mark intervals of maximum likelihood density at 95% for each estimated date that corresponds to the average obtained through Bayesian analysis for each patient separately.



