
Hereditary cancer
Why detection is crucial
ABSTRACT
Although just 1% of cancer cases can be considered hereditary, there are currently over 200 described syndromes characterised by familial aggregation of various tumour types. During the twentieth century, many of the genes underlying these syndromes have been identified, leading to a breakthrough in the management of affected families when it comes to identifying whether individuals are at risk or not, and establishing specific preventive measures. However, there is a high percentage of inherited cases for which the cause of predisposition is unknown, making the search for new genes, using the newly available technology, a research priority.
Keywords: hereditary cancer, inheritance patterns, BRCA1, BRCA2, hereditary ovarian and breast cancer syndrome.
HISTORICAL INTRODUCTION
To date over 200 hereditary syndromes associated with an increased risk of cancer have been identified. These are characterised by the occurrence of primary malignant neoplasia in numerous members of the same family, which are sometimes associated with congenital abnormalities. It was precisely the observation of these congenital anomalies affecting family groups that also led to observe the first cases of hereditary cancer. Among these are cases of neurofibromatosis type 1 (NF1), characterised by the presence of skin lesions, like «café au lait» spots and small nodules on the skin, as well as an increased risk of developing tumours in the central nervous system.
In 1866, the French surgeon Paul Broca first published an example of hereditary breast cancer, in which he described four generations of women affected by breast cancer in his wife’s family. The twentieth century witnessed numerous clinical descriptions of familial cancer clusters; however, the two milestones marking a new era in the field of hereditary cancer research were Knudson’s theory in the early seventies (Knudson, 1971) and the discovery, in 1986, of the first cancer-susceptibility gene, Rb, involved in familial retinoblastoma (Friend et al., 1986.

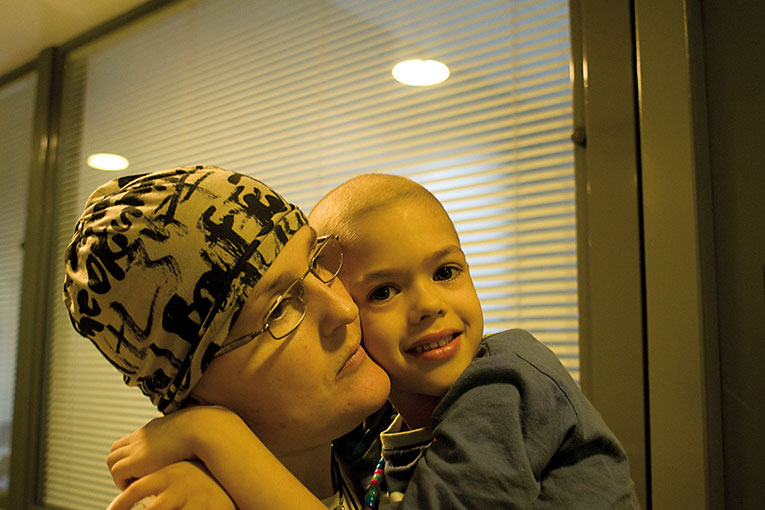
Images taken from Tino Soriano’s «David versus Goliath» series, where the artists portrays cancer, particularly childhood cancer In the picture, Sant Pau Hospital (Barcelona, Spain). / Tino Soriano
Knudson’s theory was based on the observation of 48 cases of retinoblastoma, a malignant tumour of the retina. In 1971, Knudson developed his two-hit theory, based on the observation that hereditary retinoblastoma cases developed at a much earlier age than sporadic cases (with no family history). According to Knudson’s hypothesis, the diseases is caused by two mutational events, two-hits, which would affect both copies of the gene underlying the disease. In the hereditary form, a mutation (alteration in genetic information) would be in the germline, transmitted form generation to generation, making carriers much more susceptible to developing cancer. This susceptibility is inherited in an autosomal dominant way, i.e., an abnormal gene from one parent is enough to cause susceptibility to the disease, even though the second copy of the gene inherited from the other parent is normal. The second copy alters the at the somatic level, in this case the retina, and that is when the tumour forms (Figure 1). In non-hereditary forms, both mutations occur at the somatic level.
Shortly afterwards, it was reported that this model could be extended to Wilms’ tumour (kidney tumour in childhood), infant neuroblastoma (nervous system embryonic tumour), pheochromocytoma (tumour of the adrenal gland) and, in general, all hereditary cancers. This theory has held up to the present day and, although there are exceptions, most of the major hereditary cancer syndromes follow this pattern.
PRINCIPALS GENES INVOLVED: MODELS OF INHERITANCE
Until the late twentieth century, research into inherited susceptibility to cancer focused on the identification of high penetrance or high susceptibility genes. Besides those already mentioned, other classic examples of genes are: the TP53,gene, identified in 1990 and responsible for Li-Fraumeni syndrome; the APC, gene, identified in 1991, responsible for familial colorectal polyps and cancer; or gene MSH2, discovered in 1993, involved in hereditary non-polyposis colon cancer (HNPCC). Research in this field was especially fruitful in 1994, as three of the most important genes were identified: MLH1, the second gene involved in HNPCC, BRCA1, the first gene identified to play a role in hereditary breast and ovarian cancer syndrome (HBOC), and p16, involved in familial melanoma. These are some of the most important. Finally, another noteworthy discovery was BRCA2, in 1995, the second gene predisposing carriers to breast and ovarian cancer. We will use this syndrome as an example to introduce other concepts in this article. The origin of cancer lies in the ability of certain cells to escape the mechanisms regulating normal cell growth, thus leading to their uncontrolled proliferation. All the genes mentioned so far, and most of those involved in hereditary cancer, are known as tumour suppressor genes (TSG). In general, the function of these genes is to exert a negative control on the cell cycle, preventing uncontrolled cell proliferation. There are some exceptions to this rule, in which the gene involved is not a TSG but a proto-oncogene, i.e., a gene which activates cell growth and division, such as the RET, proto-oncogene gene, involved in multiple endocrine neoplasia type 2 (MEN2). The latter differs from the TSG in that it plays a role in cell-cycle activation, and suffers activating-function mutations, whereby only one copy of the mutant gene is enough to produce a tumour.
«In 1866, the French Surgeon Paul Broca first published an example of hereditary breast cancer, in which he described four generations of women afected by breast cancer»
All the cases mentioned so far follow the pattern of autosomal-dominant inheritance; however, we must also mention some hereditary cancer syndromes that follow an autosomal-recessive pattern. In these cases, both copies of the gene must be mutated at germline level for the disease to develop. Most of the genes involved in these syndromes are involved in DNA-repair processes, which is why chromosomal instability is a common feature of such syndromes. A good example is Fanconi anaemia (FA), a disease characterised by progressive bone marrow failure, hypersensitivity to DNA-intercalating agents (molecules that are interspersed in the bases that make up DNA and affect its structure) and increased susceptibility to cancer.
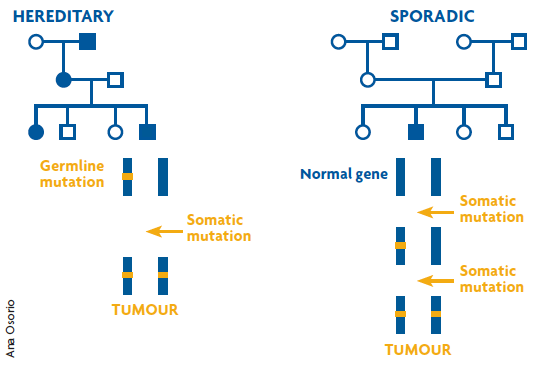
Figure 1. Knudson’s two-hit model. The figure shows two genealogical trees in which the circles represent females and the squares males, with individuals diagnosed with cancer depicted in blue. In the hereditary case (on the left), one copy of the gene responsible for the disease is mutated in the germline, this event means it is highly probable that a second somatic mutation will take place, and therefore the risk of developing cancer is very high and at an earlier age of onset. In the sporadic case (on the right) both mutations have to occur at the somatic level, which is a much more unlikely event. This explains why there is only one case of cancer in this family and the age of onset is later. / Ana Osorio
CLINICAL MANAGEMENT OF FAMILIES WITH HEREDITARY CANCER
We should point out that those cancers considered as clearly hereditary, represent only 1% of overall cases, which most often appear sporadically and whose etiology involves a host of factors.
Nonetheless, although only a small percentage of cases can benefit, identification of these high-risk genes represents a major advance in the management of families with hereditary cancer. This is because it provides the possibility to perform genetic screening of family members to identify whether they are at risk or not. Currently the only options existing to cure a patient of cancer are early detection or prevention. In this respect, the possibility of identifying a family with hereditary cancer causing predisposition enables personalised follow up of those family members who carry a particular mutation, and whom we know have a very high risk of developing cancer during their lifetime.
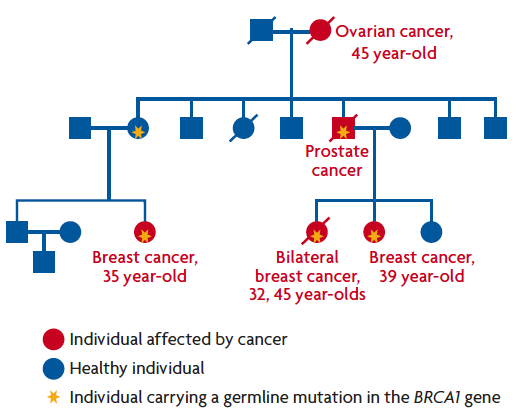
Figure 2. Example of a family with hereditary breast cancer, carrying a germline mutation in the BRCA1gene. Characteristics common to most familial cancers can be observed, such as numerous people in the same family are affected; tumours develop at an earlier age than the average age at which tumours are diagnosed in the general population; bilateral presence in the case of cancer affecting organs present in pairs, and associated tumours, as in the case of ovarian cancer. / Ana Osorio
Let us look, for example, at a family with hereditary breast and ovarian cancer (Figure 2). Once the mutation in BRCA1 which is responsible for susceptibility, has been detected, all women in the family who have not developed cancer can be screened. Those who are found to carry the gene are at higher risk of developing cancer, and can thus receive personalised monitoring programmes, which will detect the tumour at an early stage when there is a higher probability of cure. In these women at risk, depending on personal circumstances, there is even the possibility of prophylactic surgery to reduce the chance of developing breast or ovarian cancer by up to 95%. Depending on the type of hereditary tumour, detection of the mutation responsible for susceptibility may allow more or less effective measures to be taken.
For example, MEN2A syndrome is characterised by the appearance of endocrine tumours, in particular medullary carcinoma of the thyroid, pheochromocytoma and parathyroid hyperplasia, and is caused in most cases by inherited mutations in the RET proto-oncogene mentioned above. The nature of this gene makes individuals carrying mutations develop a tumour in 100% of cases, as it is only necessary for one copy of the gene to be altered, which is an exception to Knudson’s two-hit model. The main primary preventive intervention measure is prophylactic thyroidectomy in childhood, which completely eradicates the risk of developing medullary thyroid cancer.
At the opposite extreme we find syndromes associated with multiple tumours, such as Li-Fraumeni syndrome, characterised by the appearance of soft tissue sarcomas, osteosarcomas, breast cancer, brain tumours and leukaemia. In this syndrome patient follow-up is much more complicated, not only due to the likelihood of developing different types of tumour but because some of them are very difficult to diagnose early on.
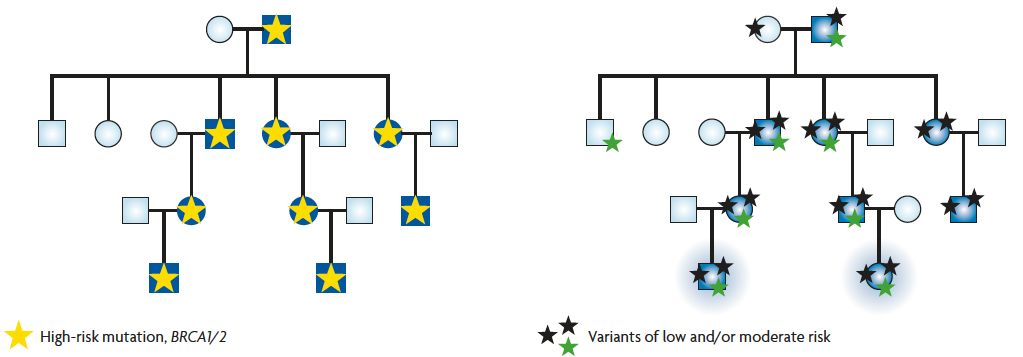
Figure 3. The genealogical tree on the left represents a classic case of hereditary cancer in which all affected individuals of the family (in blue) are explained by a mutation in a high-risk gene (yellow star). The genealogy on the right represents the same family, but this time, cancer cases are due to the accumulation and combination of variants of low and/or moderate risk. Cancer will only develop in cases where a suffi cient number of variants accumulates whereas those with only one variant will not develop cancer, as the risk conferred individually is very low. / Adapted from Cirulli and Goldstein, 2010
OTHER CANCER-SUSCEPTIBLE GENES
One of the main problems arising when dealing with hereditary cancer is not the detection of the mutation causing susceptibility. In most hereditary cancer syndromes, there are some cases that cannot be explained by mutations in known genes, one of the best examples of this problem is the HBOC syndrome.
«The origin of cancer lies in the ability of certain cells to escape the mechanisms regulating normal cell growth, thus leading to their uncontrolled proliferation»
When the BRCA1 and BRCA2 genes were identified almost twenty years ago, it was thought that the germline mutations identified could explain up to 80% of cases of hereditary breast cancer, but a few years later it was confirmed that this percentage was not so high and could be variable depending on the population tested. Currently it is considered that, in general, these two genes do not account for more than 20% of cases, which has meant that for the last decade the search for new susceptibility genes has been one of the main research goals. However, since the 1990s no other high-risk gene specifically involved in the syndrome has been identified. This has led to the acceptance of a polygenic model, which would explain the increased risk and family grouping, whereby the combination of low- or moderate-risk variants would accumulate in these families (Figure 3).
Individually, each variant would alter the risk very little, but in conjunction with other factors, both genetic and environmental, these genes with low or moderate penetrance would not only be involved in these family patterns but would also be responsible for differences in cancer susceptibility between individuals in the general population. In the last five years, Genome Wide Association Studies, commonly known as GWAS, have led to a breakthrough in the discovery of some of these variants (Visscher, 2012). This model is also being applied to other types of hereditary cancer.
Bibliography
Cirulli, E. T., & D. B. Goldstein, 2010. «Uncovering the Roles of Rare Variants in Common Disease Through Whole-genome Sequencing». Nature Reviews Genetics, 11: 415-425. DOI: <10.1038/nrg2779>.
Friend, S. H. et al., 1986. «A Human DNA Segment with Properties of the Gene that Predisposes to Retinoblastoma and Osteosarcoma». Nature, 323: 643-646. DOI: <10.1038/323643a0>.
Knudson Jr., A. G., 1971. «Mutation and Cancer: Statistical Study of Retinoblastoma». PNAS, Proceedings of the National Academy of Sciences, 68(4): 820-823. DOI: <10.1073/pnas.68.4.820>.
Ponder, B. A.; Antoniou, A.; Dunning, A.; Easton, D. F., & P. D. Pharoah, 2005. «Polygenic Inherited Predisposition to Breast Cancer». Cold Spring Harbor Symposia Quantitative Biology, 70: 35-41. DOI: <10.1101/sqb.2005.70.029>.
Visscher, P. M.; Brown, M. A.; McCarthy, M. I., & J. Yang, 2012. «Five Years of GWAS Discovery». The American Journal of Human Genetics, 90: 7-24. DOI: <10.1016/j.ajhg.2011.11.029>.




