
If one thing has become clear in the current COVID-19 pandemic, it is that everything has happened at breakneck speed, and science has been no exception. Since the authorities of the Republic of China informed the World Health Organisation on 31 December 2019 that they were treating dozens of cases of pneumonia of unknown origin, scientists from all over the planet have been working to first discover the pathogen hiding behind these pneumonias and then to study it in detail in order to develop a therapeutic strategy to mitigate its devastating effects.
Thus, in the first weeks of January a group of researchers from Fudan University (China) sequenced the genome of SARS-CoV-2, the coronavirus responsible for the new disease, from samples of the bronchoalveolar lavage fluid of a 41-year-old patient with no clinical history of hepatitis, tuberculosis, or diabetes, and released it even before it was accepted for publication on 28 January (Wu et al., 2020). Since then, researchers around the world have been racing to understand the molecular details of this pathogen. Today, just a few months later and with several thousand genomes sequenced, a huge amount of information has been generated that is certainly difficult to handle.
Genome organisation and expression
The SARS-CoV-2 genome consists of single-stranded RNA of about 29,900 nucleotides that can be recognised and translated by the protein synthesis machinery of infected cells. This genome can encode up to 29 proteins, although it is not yet clear whether all of them are actually synthesised during the life cycle of the virus. Four of the proteins encoded in the genome shape the coronavirus and are therefore known as structural proteins. These four structural proteins are the surface glycoprotein (Spike, S) that forms the characteristic spicules of this family of viruses; the protein that forms the membrane matrix (Membrane, M) which is the most abundant in the lipid envelope of the virus; a small protein also embedded in the lipid membrane that is found in a small number of copies (Envelope, E), and the protein that associates with viral RNA, stabilizing it and forming the virus nucleocapsid (Nucleocapsid, N).
«If one thing has become clear in the current COVID-19 pandemic, it is that everything has happened at breakneck speed, and science has been no exception»
These four structural proteins are encoded at the 3′ end of the genome (Figure 1) and, like nine other accessory proteins encoded in this region, are not translated directly by the ribosomes of the infected cell, but rather are synthesised from subgenomic RNA (sgRNA) generated by a viral protein responsible for replicating (copying) the virus genome (nsp12, see below). At present we still do not know if all these accessory proteins are synthesised in CoVid19 patients, and in fact the most recent studies have not managed to detect all sgRNAs (Kim et al., 2020) and there is also no evidence of the presence of some of these proteins in infected cells.
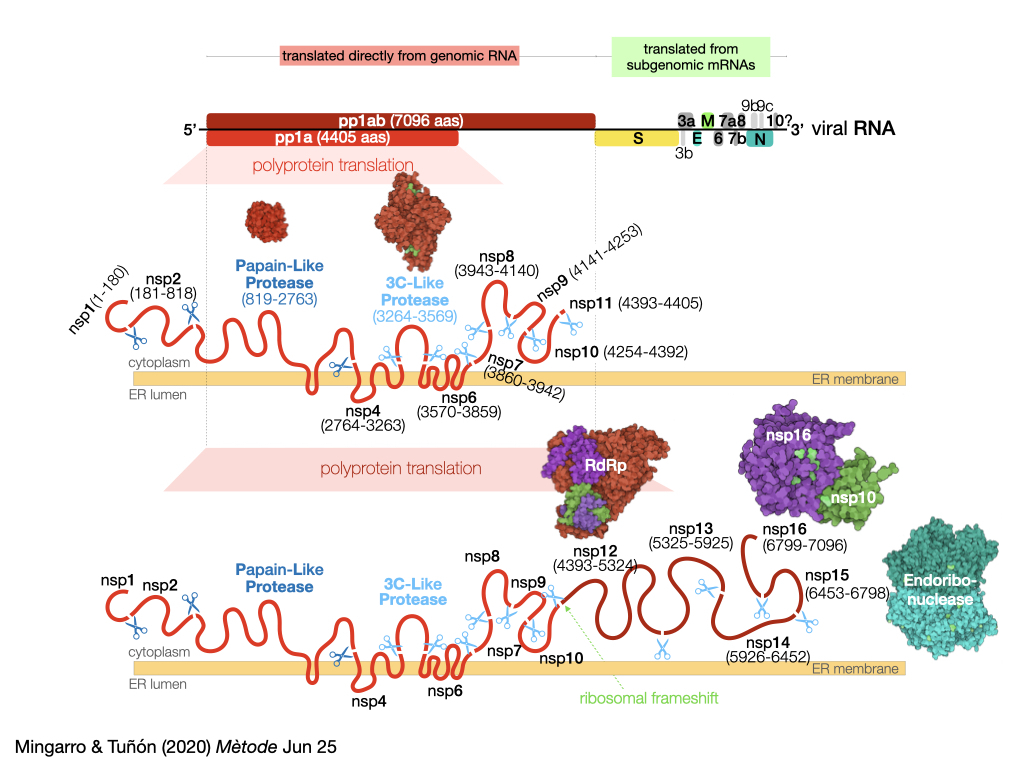
Figure 1. Schematic representation of the organisation of the SARS-CoV-2 genome and the proteins it encodes, illustrating in greater detail the pp1a and pp1ab polyproteins containing the PL and 3CL proteases.
Unlike structural and accessory proteins, which need sgRNAs as ‘messengers’ for their synthesis, the rest of the coronavirus genome encodes, at its 5′ end (this is not accidental), two polyproteins that can be synthesised directly from genomic RNA by the ribosomes of the infected cell. This means that the region, which represents approximately 67 % of the virus genome, is organised as two open reading patterns (ORF1a and ORF1ab, see Figure 1) that encode several proteins that are not separated from each other, but are synthesised as long chains of amino acids that must be digested (cut) to generate 16 non-structural proteins (nsp). As we shall see, this peculiarity in the viral life cycle constitutes one of its potential Achilles’ heels.
The coding regions of ORF1a and ORF1ab share the same information at their 5′ end, so when the ribosomes bind to the viral RNA they synthesise the membrane polyprotein 1a (pp1a) in the endoplasmic reticulum of the infected cell. As we have pointed out, this large polyprotein of 4405 amino acids must be cut to produce the functional proteins of the virus. To do this, the chemical bonds – peptide bonds – that make up the skeleton of the proteins must be broken. Under normal conditions this process is extremely slow, which explains why the proteins are stable in living cells (the time needed for the peptide bonds to break at room temperature is several hundred years).
«In order to develop effective antiviral drugs, it is necessary to have detailed knowledge of the structure and action mechanism of proteases»
To perform this digestion at a speed compatible with the replication cycle of the virus, the action of enzymes, proteins specialised in accelerating chemical reactions, is necessary. The virus encodes two of these proteins, which are named after the function they perform: proteases, protein breakdown. The most important of these proteases, 3CL (3Chymotrypsin-Like), is part of pp1a and once synthesised it self-proteolyses (cuts itself) and in its mature form makes 5 additional cuts in the polyprotein sequence to generate the non-structural proteins nsp4-nsp11. In addition to this protease, coronaviruses encode another one called PL (Papain-Like), which hinders the cellular response to the virus and makes 3 cuts in the pp1a, generating the nsp1, nsp2, and itself.
The end of the sequence coding pp1a has a stop signal (a stop codon) that indicates the end of the polyprotein synthesis to the ribosome. However, in approximately 60 % of cases, the ribosomes can undergo a change in the reading pattern (ribosomal frameshift) when this region of the viral RNA is translated, and the stop signal disappears. This change allows ribosomes to read beyond the region encoded by pp1a, leading to the synthesis of pp1ab. Pp1ab is a huge polyprotein (7096 amino acids) that includes, in addition to the non-structural proteins described in pp1a and among others, the enzyme that copies the virus genome (RNA-dependent RNA polymerase, nsp12), which we discussed earlier.
Inhibiting proteases
As we can imagine, the activity of both proteases is crucial for virus replication, so they have emerged as one of the preferred targets for the development of drugs to treat this terrible disease. In order to develop effective antiviral drugs, it is necessary to have detailed knowledge of the structure and the mechanism of action of the proteases. Again in record time (in January!) we had the first high-resolution structures of the 3CL protease (Jin et al., 2020a; Zhang et al., 2020). The swiftness of this remarkable success by two independent research groups (one in Berlin and one in Shanghai) was largely due to the experience that both groups gained working with the SARS-CoV coronavirus, which caused the SARS epidemic that broke out in Southeast Asia between 2002 and 2004. The proteins of SARS-CoV-2 are very similar to those of this other coronavirus (up to 96 % in the case of the main protease) and to those of the coronavirus causing the outbreak of the Middle East Respiratory Syndrome (MERS) in 2012.
It is rather interesting that lines of research projects that might have been considered irrelevant just a few months ago because both outbreaks mentioned above were controlled, have now become key to address the SARS-CoV-2 study quickly. This fact warns us, once again, of the danger of scientific research being guided solely by criteria of apparent usefulness or immediate applicability.
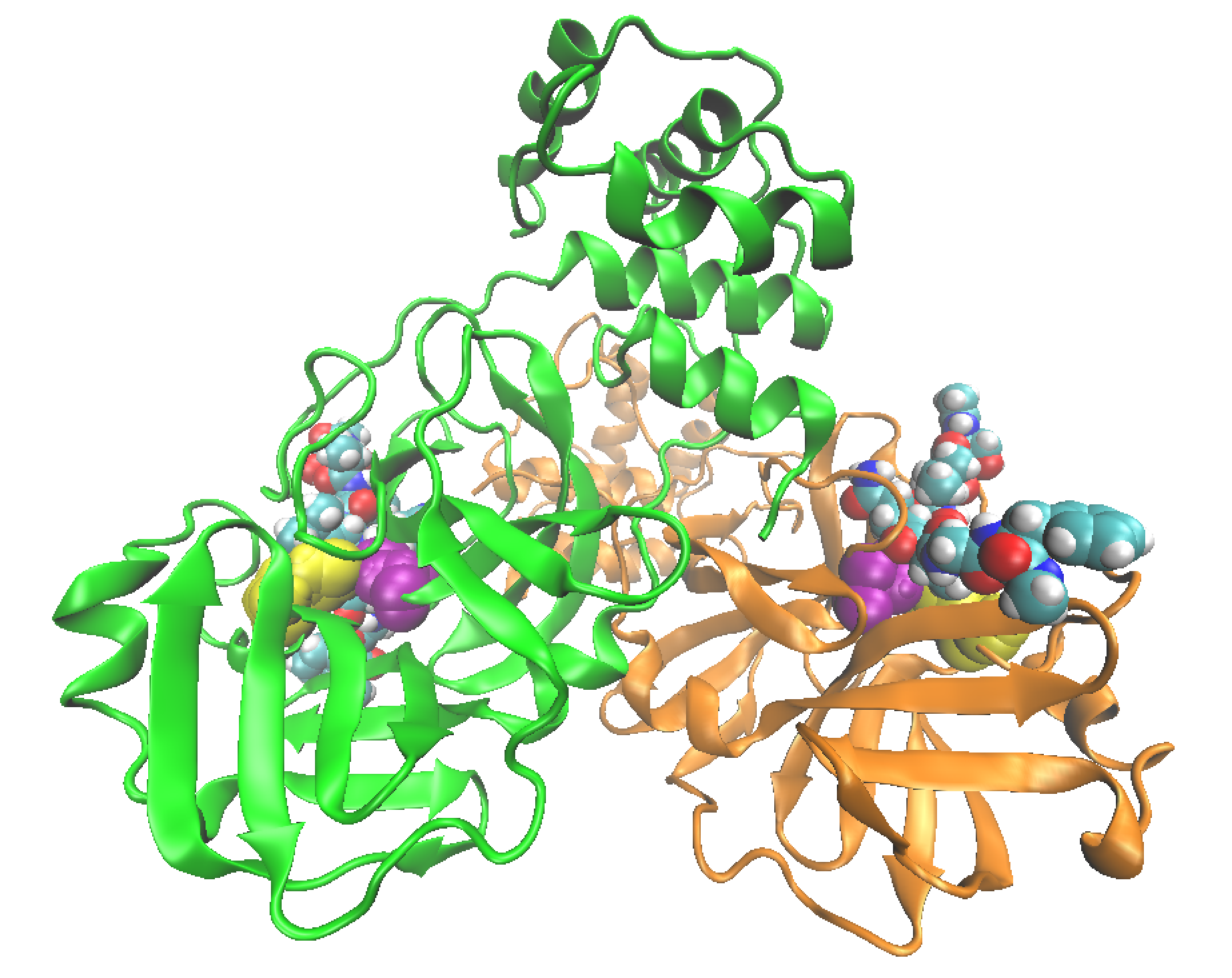
Figure 2. Representation of the structure of the 3CL-Like protease. Each of the protomers is represented in a colour with a substrate peptide (represented as spheres for each of its atoms) in each of the active centres. Catalytic cysteine and histidine are represented as spheres in yellow and purple, respectively, in each of the protomers.
The 3C-Like protease of the virus is a dimer, formed by two identical subunits, as can be seen in Figure 2. The formation of the dimer is essential for its activity, since the active site, the specific place where the protease joins the polyprotein substrate and carries out its activity, is completely formed when the interaction between the two subunits occurs. Each of the two active sites of the dimer contains a pair of amino acids responsible for breaking peptide bonds in the polyproteins, the catalytic diad formed by the Cys145 cysteine and the His41 histidine (see attached video).
The main protease of the virus is highly specific in breaking the peptide bond formed between a glutamine residue and another small amino acid (glycine, alanine, or serine). None of the proteases known in humans present the same sequence specificity, which makes the main protease of SARS-CoV-2 an excellent therapeutic target, as a drug designed to inhibit its activity will have a low probability to interact with human proteases and therefore of having side effects. These inhibitors are molecules capable of binding to the active centre of the protease, preventing access to the polyproteins and thus impeding the protease from performing its function, which implies interrupting the virus replication cycle. In fact, inhibition of major virus proteases is already an efficient strategy used for the treatment of other epidemics, such as AIDS.
As noted above, the design of inhibitors for SARS-CoV-2’s 3C-Like protease is being carried out using the knowledge gained about inhibiting the equivalent (homologous) enzyme from SARS-CoV. The Berlin group (Zhang et al., 2020) used the structural data obtained for the SARS-CoV-2 major protease to optimise an inhibitor that had already been shown to be effective against the SARS-CoV and MERS-CoV proteases. This inhibitor is capable of irreversible binding to the active site of the protease, forming a stable bond with the catalytic cysteine, thus rendering it useless for performing its function on polyproteins.
Meanwhile, the Shanghai group obtained the structure of the main protease together with a different inhibitor also linked to catalytic cysteine (Jin et al., 2020a). This inhibitor was also developed against the main proteases of SARS-CoV and MERS-CoV. In addition, the team in Shanghai conducted a screening of known drugs and natural products with the aim of selecting other possible starting points for developing more effective inhibitors. They selected 30 candidates whose protease complex structures are being resolved at the Diamond Light Source, a synchrotron laboratory in the UK. This laboratory has launched an interesting initiative by making all the solved protease-inhibitor complex structures accessible to the public and opening a website where it is possible to suggest possible inhibitors, which are tested after being evaluated by artificial intelligence, prioritising them based on criteria such as ease of synthesis or possible toxicity.
«Obtaining a drug capable of inhibiting any of the proteases in SARS-CoV-2, or any other pathogenic agent, is a long and complex process that requires a truly multidisciplinary approach»
The PL protease (Papain-Like) also contains in its active centre a cysteine and a histidine responsible for breaking the peptide bond. Therefore, the mechanism used to act as a protease is very similar to that used by 3C-Like. However, the PL protease cleaves specifically peptide bonds located after two consecutive glycines, using a recognition sequence that, unfortunately, is also used by some human proteases. This recognition sequence coincidence makes it more difficult to design specific inhibitors towards PL protease that do not, in turn, cause side effects by also interfering with the activity of human proteases. Here again, studies of the SARS-CoV or MERS-CoV PL protease, which are very similar to those of SARS-CoV-2, may facilitate the development of protease-specific inhibitors.
Despite all these achievements in such a short period of time, we must stress that obtaining a drug capable of inhibiting any of the proteases in SARS-CoV-2, or any other pathogenic agent, is a long and complex process that requires a truly multidisciplinary approach. The process can begin by determining the structure of the target protein, followed by the computer-assisted design of candidate molecules to inhibit its activity, chemical synthesis, biochemical in vitro activity measurements and, finally, the study of its viability as a drug administered first to cells, then to experimental animals and finally to humans. This last step is essential to check the possible side effects that the inhibitor may cause on our organism.
The timeframes can be significantly shortened if the inhibitor found is an already approved drug to treat other diseases. This is the reason why massive in vitro and in vivo tests are being carried out on different drugs to inhibit some of the SARS-CoV-2 proteins, including protease. Thus, the Shanghai group (Jin et al., 2020b) has recently published a structure of the main protease of SARS-CoV-2 inhibited by carmofur, an antineoplastic drug used to fight colorectal cancers and which could be a promising lead compound for the development of new antivirals. When exactly they will be available, only science (and its funding) will tell.
Representation of the Sγ nucleophilic attack (in yellow) of cysteine-145 of the 3CL protease on the peptide bond of the polyprotein substrate (the direction of the attack has been highlighted as a dashed line). Previously, histidine-41 transferred a hydrogen atom (represented in white with a dashed line) to the nitrogen atom of the peptide bond to be broken. The video is courtesy of Carlos A. Ramos-Guzmán, researcher at the University of Valencia.
Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., ... Yang, H. (2020a). Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature, 582, 289–293. doi: 10.1038/s41586-020-2223-y
Jin, Z., Zhao, Y., Sun, Y., Zhang, B., Wang, H., Wu, Y., ... Rao, Z. (2020b). Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nature Structural & Molecular Biology, 27, 529–532 (2020). doi: 10.1038/s41594-020-0440-6
Kim, D., Lee, J. Y., Yang, J. S., Kim, J. W., Kim, V. N., & Chang, H. (2020). The architecture of SARS-CoV-2 transcriptome. Cell, 181(4), 914–921. doi: 10.1016/j.cell.2020.04.011
Wu, F., Zhao, S., Yu, B., Chen, Y. M., Wang, W., Song, Z. G., ... Zhang, Y. Z. (2020). A new coronavirus associated with human respiratory disease in China. Nature, 579, 265–269. doi: 10.1038/s41586-020-2008-3
Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., ... Hilgenfeld, R. (2020). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science, 368(6489), 409–412. doi: 10.1126/science.abb3405